

新しく研究室を立ち上げるとき、限られた予算の中でどこまで環境を整えれば安全性や品質基準を満たせるのか、不安に思う方は多いのではないでしょうか。この記事では、研究室予算での環境整備を成功させるための考え方や具体的な手順、予算別の整備例までわかりやすくご紹介します。
研究室予算での環境整備の結論|優先順位を決めれば低予算でも実現できる
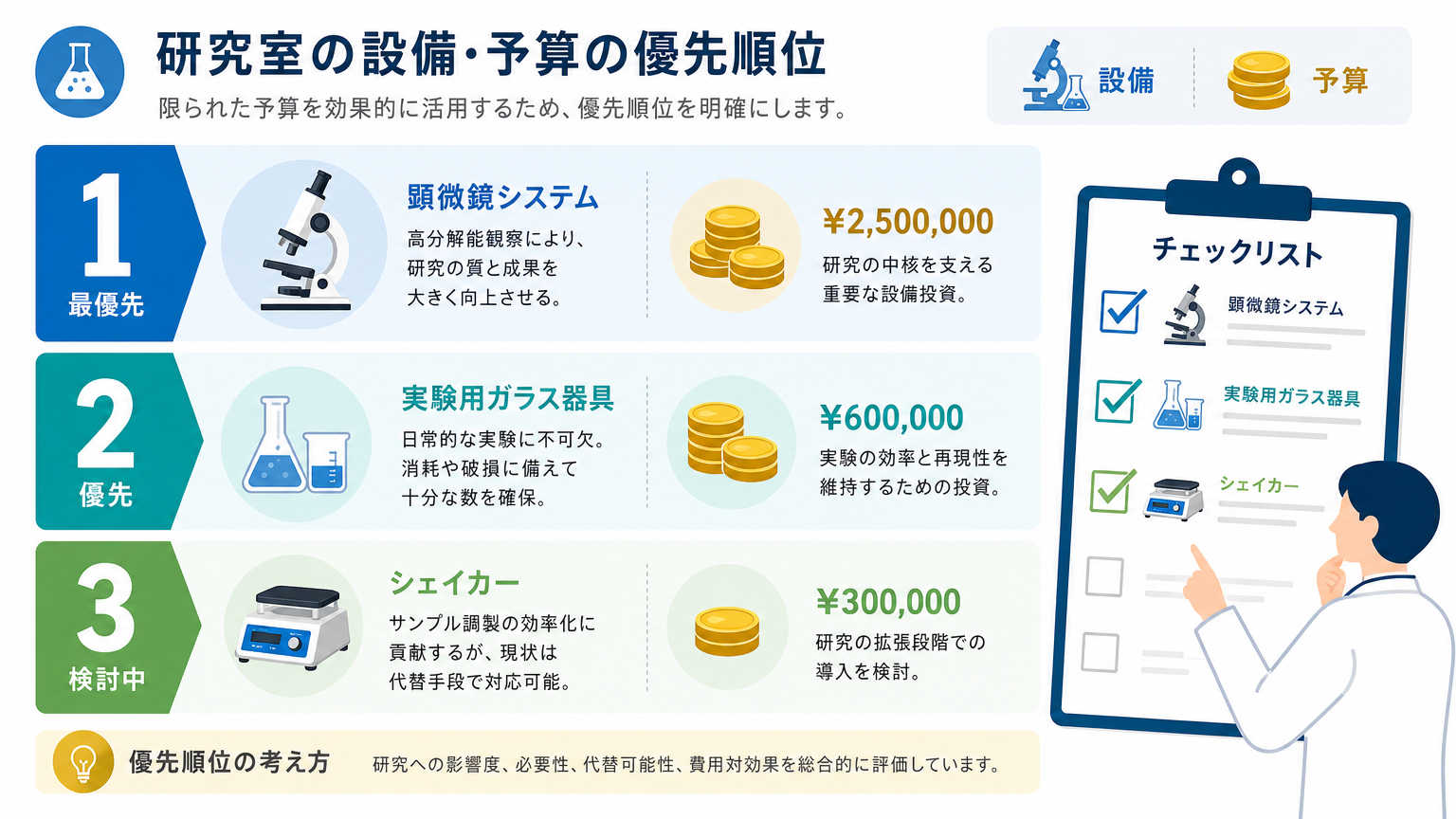
研究室予算での環境整備は、すべての設備を一度にそろえる必要はありません。安全性や品質基準に直結する必須設備を先に確保し、後回しにできる設備を分けることで、限られた予算でも十分な環境をつくれます。
結論から言うと、優先順位づけと助成金の活用、中古設備やリースの併用という3つの取り組み方を組み合わせることが、低予算でも失敗しない環境整備の近道です。まず何が「絶対に必要」で、何が「あとから追加できるもの」なのかを整理するところから始めましょう。
研究の種類や規模によって必須設備は変わりますが、細胞培養や再生医療関連の研究室であれば、清浄度を保つための設備や温度管理された保管環境は優先度が高くなります。逆に、作業効率を上げるための付属設備は、予算に余裕ができた段階で追加しても問題ないケースがほとんどです。
次の章では、なぜ環境整備を後回しにできないのか、その理由を具体的に見ていきます。予算の制約と安全基準のバランスをどう取るべきか、順番に確認していきましょう。
限られた予算でも環境整備が必要な理由
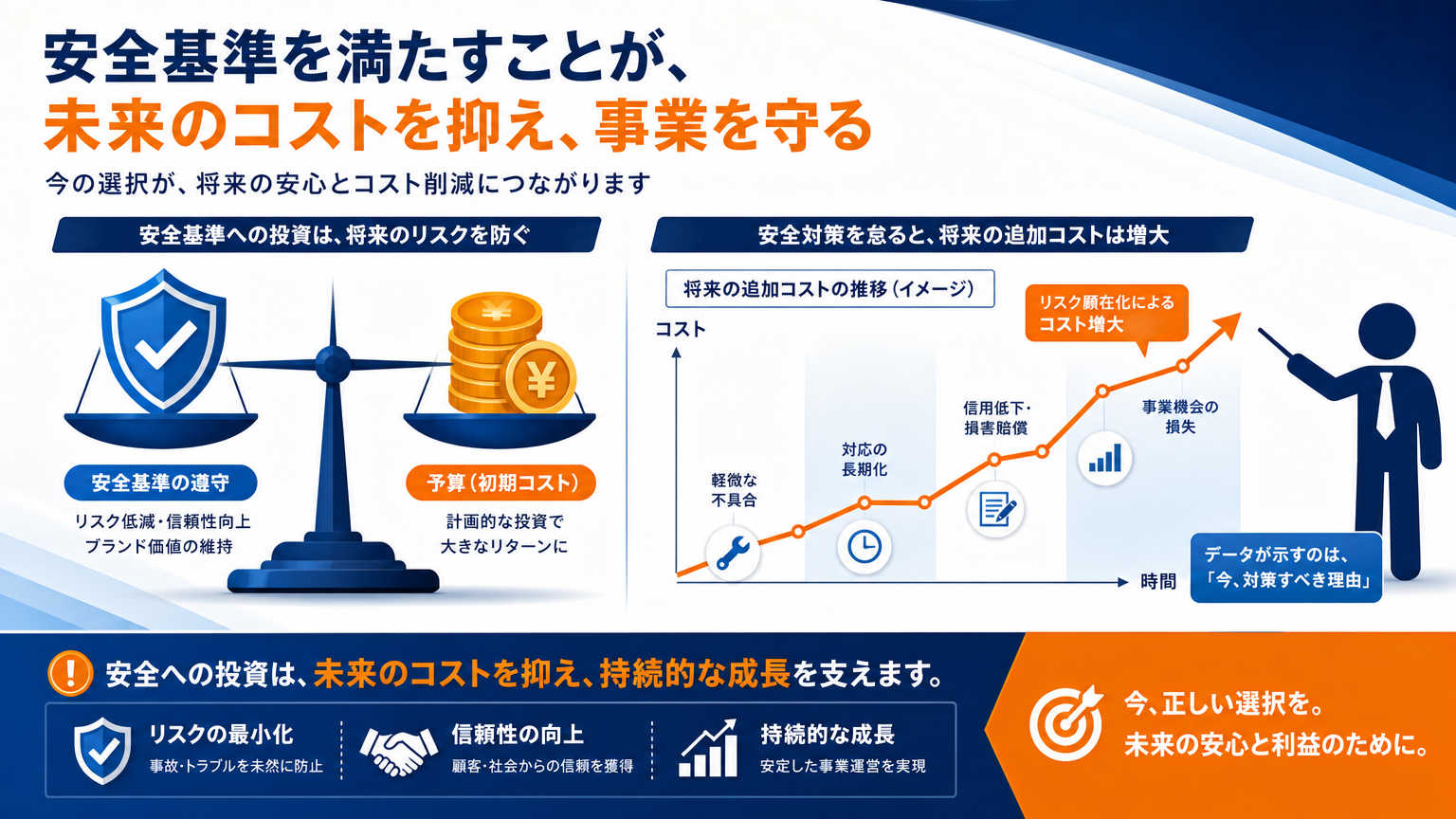
研究室の環境整備は、単なる見栄えや作業効率の問題ではありません。安全基準や品質基準を満たせなければ研究自体が止まってしまいますし、初期投資を惜しむと後で余計な費用がかかることもあります。ここでは2つの視点から、環境整備を先送りできない理由を説明します。
安全基準や品質基準を満たさないと研究や実験が続けられないから
細胞培養や再生医療の研究では、清浄度や温度管理などの基準を満たさないと、実験データの信頼性そのものが損なわれてしまいます。たとえば培養環境に微生物汚染のリスクがあると、せっかくの実験結果が使えなくなることもあるでしょう。
再生医療等製品の製造では、GMP(Good Manufacturing Practice)に準じた施設要件が求められる場合があります。基準を満たさない環境で実験を続けても、後から得られたデータが評価されず、研究計画全体が見直しになるリスクがあります。
予算が限られているからといって基準を無視してしまうと、結果的に研究の信頼性を失い、時間とお金の両方を無駄にしてしまいます。だからこそ、少ない予算の中でも基準を満たす部分から優先的に整えることが大切です。
立ち上げ初期に環境整備を後回しにすると後で費用がかさむから
「今は予算がないから、あとで整備すればいい」と考えてしまう方もいるかもしれません。ですが、後から設備を追加する場合、配線や配管の工事をやり直したり、既存のレイアウトを変更したりする手間が発生し、結果的に初期投資よりも高くつくことが少なくありません。
特に空調設備や電源容量などの基盤部分は、後から変更しようとすると大がかりな工事になりがちです。最初の設計段階で将来的な拡張を見込んでおくだけでも、後々の追加コストをかなり抑えられます。
立ち上げ時にすべてをそろえる必要はありませんが、「将来どこまで拡張する可能性があるか」を見据えて基盤部分だけは先行して整えておくのが賢い進め方です。
研究室予算での環境整備で失敗しないための考え方
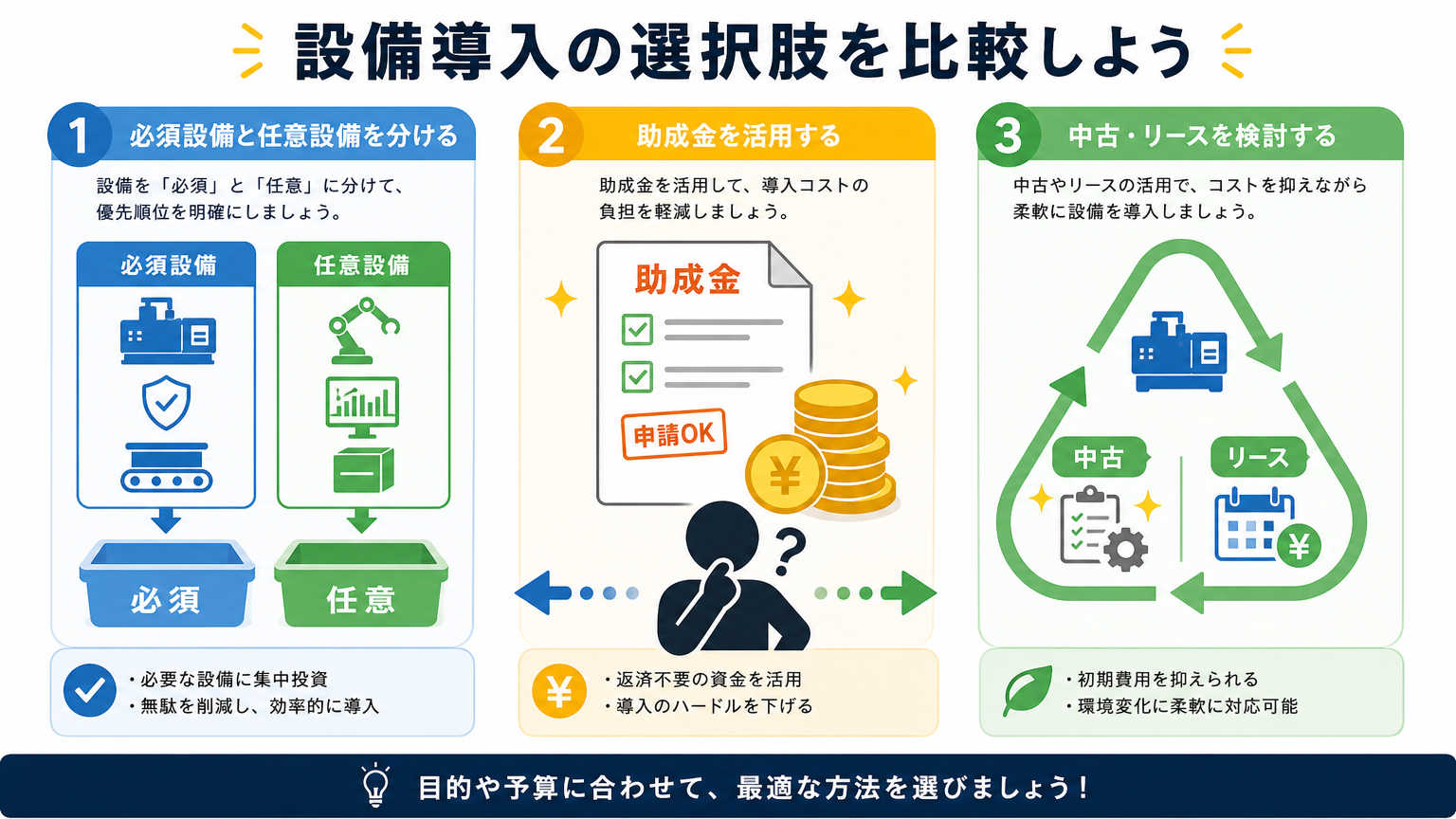
予算内で環境整備を進めるには、闇雲に設備を買い集めるのではなく、いくつかの考え方の軸を持つことが重要です。ここでは優先順位づけ、助成金の活用、そして中古やリースの活用という3つの視点から、失敗しない進め方を紹介します。
必須設備と後回しにできる設備を分ける
環境整備でよくある失敗は、必要な設備と欲しい設備の区別があいまいなまま予算を使ってしまうことです。まずは研究内容に対して「なければ実験が成立しないもの」と「あれば便利だが今すぐでなくても良いもの」に分けて考えましょう。
細胞培養設備であれば、CO2インキュベーターや安全キャビネットなどは基準上必須になることが多い一方、自動化された分析機器などは予算に余裕が出てから検討しても支障がないケースが多く見られます。
設備をリスト化し、必須・推奨・任意の3段階で優先順位をつけておくと、予算配分の判断がぐっとしやすくなります。この整理をしておくだけで、見積もり交渉の場でも軸を持って判断できるようになるでしょう。
助成金や補助金を活用して予算を増やす
研究室予算での環境整備は、自己資金だけでなく助成金や補助金を活用することで大きく幅が広がります。文部科学省や経済産業省をはじめ、地方自治体でも研究施設整備を対象とした支援制度が用意されている場合があります。
代表的な支援策としては、科学研究費助成事業(科研費)の設備備品費や、中小企業向けのものづくり補助金などが挙げられます。詳細な条件や申請時期は年度によって変わるため、最新情報は各制度の公式サイトで確認することをおすすめします。
申請には準備期間が必要なため、環境整備の計画を立てる段階から助成金の存在を意識しておくと、予算全体の見通しが立てやすくなります。
中古設備やリース活用でコストを抑える
新品の設備をすべて購入すると、それだけで予算の大半を使い切ってしまうこともあります。そこで検討したいのが、中古設備の購入やリース契約による導入です。
中古設備は購入価格を大きく下げられる一方、動作保証や消耗部品の状態を事前にしっかり確認する必要があります。リースであれば初期費用を抑えつつ、契約期間終了後に新しい機種へ切り替えやすいというメリットもあります。
| 導入方法 | メリット | 注意点 |
|---|---|---|
| 新品購入 | 保証が長く安心して使える | 初期費用が高い |
| 中古購入 | 費用を大きく抑えられる | 状態確認や保証範囲の確認が必要 |
| リース | 初期費用が少なく更新しやすい | 長期的には割高になる場合もある |
どの方法が合っているかは設備の種類や使用頻度によって変わるため、複数の選択肢を比較しながら検討するのが良いでしょう。
研究室予算での環境整備の具体的な手順
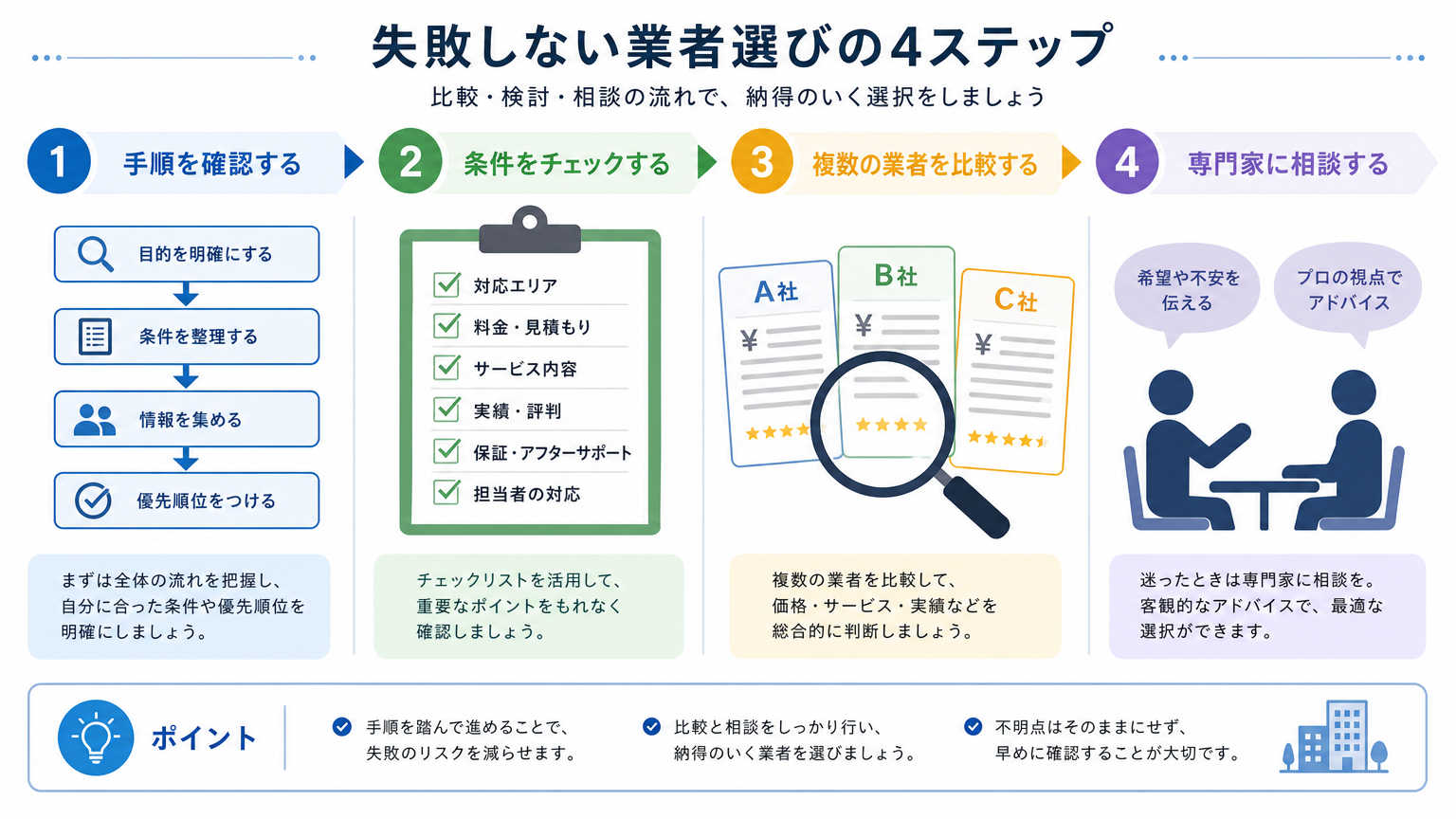
考え方が整理できたら、実際に環境整備を進める手順に移りましょう。ここでは設備のリストアップから見積もり比較、専門業者への相談まで、実際の進め方を順番に説明します。
必要な設備をリストアップして優先順位をつける
最初のステップは、研究内容に必要な設備をひとつずつ書き出すことです。培養設備、保管設備、測定機器、消耗品保管スペースなど、カテゴリごとに整理すると漏れが少なくなります。
リストアップした後は、前章で触れた必須・推奨・任意の3段階で優先順位をつけましょう。この段階で研究チームのメンバーや上司とすり合わせておくと、後になって「これも必要だった」という手戻りを減らせます。
優先順位が明確になれば、限られた予算をどこに配分するかの判断がしやすくなり、見積もり依頼の際にも要望を的確に伝えられるようになります。
複数の業者から見積もりを取り比較する
設備リストが固まったら、複数の業者から見積もりを取りましょう。1社だけの見積もりでは、価格が適正かどうかの判断がつきにくいためです。少なくとも2〜3社から相見積もりを取ることをおすすめします。
比較する際は価格だけでなく、保証内容やアフターサービス、納期なども合わせて確認しましょう。安いからといって選んだ業者が、設置後のメンテナンス対応が弱いというケースも見られます。
見積もり比較用のチェックリストを用意しておくと、抜け漏れなく条件をそろえて比較できます。
- 本体価格と設置費用の内訳
- 保証期間とアフターサポートの範囲
- 納期と設置までのスケジュール
- 消耗品や交換部品の入手性
専門業者に相談して過不足のない構成にする
設備選定は研究者だけで判断するより、施設整備に詳しい専門業者に相談したほうが、無駄のない構成にまとまりやすくなります。専門業者は法規制や基準の変更にも詳しいため、思わぬ見落としを防げます。
特に細胞治療や再生医療の分野では、清浄度管理や動線設計など専門的な知識が求められる場面が多くあります。研究期間や医療施設向けの設備に詳しい業者であれば、予算に応じた提案や将来の拡張性を踏まえた設計まで相談できるでしょう。
自分たちだけで判断に迷う部分があれば、早い段階で相談してしまうほうが結果的に手間もコストも抑えられます。
予算別に見る研究室環境整備の具体例
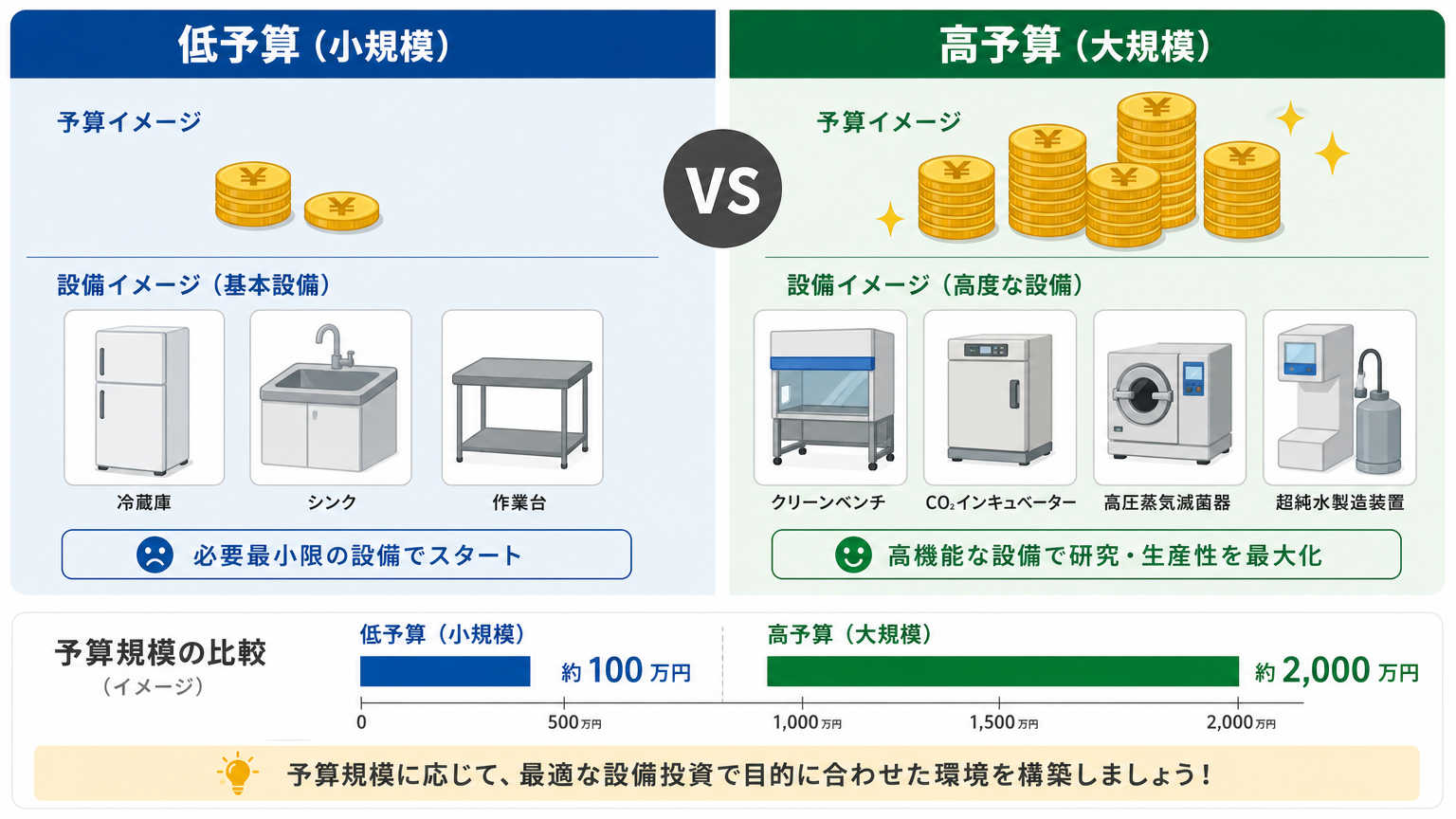
実際にどの程度の予算でどのような環境が整えられるのか、イメージしづらいという方も多いでしょう。ここでは数十万円規模と数百万円規模の2パターンで、整備できる内容の目安を紹介します。
数十万円規模でそろえる最低限の環境
数十万円規模の予算では、基本的な実験や小規模な細胞培養を行うための最低限の環境を整えることになります。中古設備やリースを活用しながら、必須設備だけを優先的に確保するイメージです。
| 項目 | 内容の目安 |
|---|---|
| 培養設備 | 中古のCO2インキュベーター、簡易型安全キャビネット |
| 保管設備 | 小型冷凍庫・冷蔵庫 |
| 消耗品管理 | 棚やラックによる整理保管 |
この段階では作業効率を高める周辺設備までは手が回らないことが多いですが、安全基準を満たす部分に予算を集中させることで、最低限必要な研究環境は確保できます。予算が増えた段階で、少しずつ設備を拡充していく計画を立てておくと良いでしょう。
数百万円規模で整える標準的な環境
数百万円規模になると、新品設備を中心にそろえながら、清浄度管理された作業スペースなどより本格的な環境整備が可能になります。細胞治療関連の研究であれば、クリーンベンチや専用の空調設備を導入できる予算感です。
| 項目 | 内容の目安 |
|---|---|
| 培養設備 | 新品のCO2インキュベーター、クリーンベンチ |
| 保管設備 | 医療用冷凍庫、温度管理された保管庫 |
| 環境管理 | 簡易的な清浄度管理設備、モニタリング機器 |
この規模になると、研究の再現性や品質を安定させやすくなり、外部評価や共同研究の際にも環境面での信頼性を示しやすくなります。ただし、設備が増える分、メンテナンス費用も増えるため、運用コストまで見込んで計画を立てることが大切です。
まとめ

研究室予算での環境整備は、必須設備と後回しにできる設備を分けることから始まります。安全基準を満たす部分を優先し、助成金や中古設備・リースを組み合わせることで、限られた予算でも十分な環境を整えられます。
手順としては、設備のリストアップと優先順位づけ、複数業者からの見積もり比較、専門業者への相談という流れを踏むことで、過不足のない構成にまとまりやすくなります。数十万円規模から数百万円規模まで、予算に応じた整備の目安を参考に、まずは自分の研究室に必要なものから整理してみてください。
研究室予算での環境整備についてよくある質問

-
研究室の環境整備で最初にお金をかけるべきものは何ですか?
- 安全基準や品質基準に直結する設備を優先しましょう。細胞培養であればCO2インキュベーターや安全キャビネットなど、基準上必須となる設備が最初に確保すべき対象です。作業効率を上げる付属設備は後回しにしても問題ないケースが多いです。
-
助成金はどのタイミングで申請すればいいですか?
- 申請には準備期間や審査期間があるため、環境整備の計画を立てる段階から情報収集を始めるのがおすすめです。年度によって公募のスケジュールが異なるため、各制度の公式サイトで最新の募集要項を確認してください。
-
中古設備を選ぶ際に注意すべき点はありますか?
- 動作保証の有無や消耗部品の残り寿命、過去のメンテナンス履歴を確認しましょう。特に温度管理が重要な設備は、精度が保たれているか事前に確認しておくと安心です。
-
見積もりは何社から取るのが良いですか?
- 少なくとも2〜3社から相見積もりを取ることをおすすめします。価格だけでなく保証内容や納期、アフターサービスの範囲まで比較すると、条件の違いが見えやすくなります。
-
予算が少ない場合、専門業者に相談しても意味がありますか?
- 予算が少ないからこそ、専門業者への相談が有効です。無駄のない設備構成や優先順位のつけ方について、専門的な視点からのアドバイスを受けられるため、限られた予算を効果的に使いやすくなります。


